The hematology world is a-twitter about the imminent approval of Zynteglo (betibeglogene darolentivec), the lentiglobin cell and gene therapy from Bluebird Bio Inc. that’s expected to become the first gene therapy to address the blood disease beta-thalassemia and the company’s first product to market. A nod for the treatment, which comprises autologous CD34 cells transduced with a lentiviral vector, lentiglobin 3305, expressing a working copy of the beta-globin gene (betaA-T87Q-globin), is expected this quarter by the European Commission after last month’s endorsement by the EMA’s Committee for Human Medicinal Products, paving the way for formal EU authorization. (See BioWorld, April 1, 2019.)
Beta-thalassemia is a blood disorder that reduces the production of hemoglobin, the protein in red blood cells that carries oxygen to cells throughout the body. Those with the condition – classified into thalassemia major (also known as Cooley’s anemia) and thalassemia intermedia – develop anemia and face increased risk of developing abnormal blood clots. Symptoms of thalassemia major, the more serious form, appear within the first two years of life and can lead to delayed development and life-threatening complications.
 Beta-thal, which results from broken beta-globin, or HBB, genes, was among the first genetic diseases characterized at the nucleotide level when early sequencing studies were conducted in the 1970s, observed Ellis J. Neufeld, faculty member at St. Jude Children’s Research Hospital in Memphis, where he also serves as executive vice president, clinical director, physician-in-chief and chair in pediatric medicine.
Beta-thal, which results from broken beta-globin, or HBB, genes, was among the first genetic diseases characterized at the nucleotide level when early sequencing studies were conducted in the 1970s, observed Ellis J. Neufeld, faculty member at St. Jude Children’s Research Hospital in Memphis, where he also serves as executive vice president, clinical director, physician-in-chief and chair in pediatric medicine.
“The mechanism of the disease itself has been known for a long time,” Neufeld told BioWorld Insight. “Every way that a beta-globin gene could possibly be broken has been discovered.”
Having that knowledge didn’t translate quickly into therapeutic products, however, in the same way that understanding of the similar mechanism underlying sickle cell disease (SCD) – where the same gene is broken in a “qualitative” rather than “quantitative” mutation as in thalassemia, Neufeld said – is just beginning to pay dividends in the clinic. (See BioWorld Insight, March 11, 2019.)
“Advances for potential gene therapies for beta-thalassemia and SCD have gone hand in hand,” explained Neufeld, who serves as an expert on hemostasis and thrombosis for the American Society of Hematology (ASH). For example, Bluebird’s betibeglogene darolentivec, formerly Lentiglobin BB-305, also is in phase I/II development in SCD.
‘It’s breathtaking technology’
Despite some similarities between the indications, beta-thal is uncommon among African Americans, a population where SDC is especially prevalent. Instead, thalassemia emerged in regions where malaria was endemic before the 20th century, including Mediterranean countries and those in North Africa and the Middle East as well as India and Central and Southeast Asia. The severe form of the disease remains rare in the U.S., with approximately 1,000 individuals affected each year, mostly in cities that embraced large waves of immigrants from affected countries. Neufeld named New York, Philadelphia, Oakland, Chicago and Boston as among the busiest treatment centers.
The global incidence of beta-thal, with “thousands and thousands of patients in the world but just a handful in the U.S.,” Neufeld said, makes Europe a savvy first move for Bluebird, of Cambridge, Mass. In addition to the challenge of enrolling participants in the U.S. into clinical trials, the absolute numbers of potential patients also are much larger in countries such as Italy – one of the first four target markets in Europe, along with France, Germany and the U.K. Together, Bluebird estimates about 2,500 individuals will be suitable for treatment in Europe following initial approval, eventually expanding to about 9,000.
Treating the underlying cause of beta-thal requires the production of enormous amounts of beta-globin, which demands a high level of transgene expression. A decade after scientists concluded that gene therapy could achieve that end, Bluebird has “figured out how to do that,” Neufeld said, with Zynteglo adding several grams per deciliter of the transgene.
“It’s breathtaking technology,” he said. “It’s really a great advance.”
Individuals who make small amounts of beta-globin – those with beta-plus (B+) thalassemia – are likely to see more reliable benefit from the lentiglobin construct than those with beta-zero (B0) thalassemia, Neufeld said. In fact, Zynteglo initially is indicated for those with transfusion-dependent beta-thal (TDT) who do not have a B0/B0 genotype and for whom hematopoietic stem cell transplant is appropriate but a matched related donor is not available.
In a research note earlier this month, J.P. Morgan analyst Cory Kasimov observed that “the approval of Zynteglo in non-B0/B0 [genotype] B-thal is seemingly a forgone conclusion in Europe, and we are equally confident in the U.S. based on data to date. Similarly, our conviction in SCD is materially higher following recent updates.”
During the company’s analyst day presentation this month, Bluebird execs expressed optimism about patient and physician awareness in the EU advance of the Zynteglo EU launch.
“We’ve been present in these four countries for quite a while,” Andrew Obenshain, head of Europe and senior vice president for Bluebird, explained during the analyst day conference call. “We have physicians who have experience with our product and can be advocates for that product. And we’ve learned a lot about which physicians treat those patients and where those patients are located.” For example, six centers in Germany see approximately half of the country’s beta-thal patients, and 15 centers in the U.K. see about 75% of patients. “Italy is a little bit of the outlier, with 73 centers,” he added, “because there are so many patients there and so many centers that see them.”
“Though the launch will be slow and on a country-by-country basis, there have been clinical trial sites in all of the initial four countries for launch, which brings a high level of [key opinion leader] insight and interest,” Kasimov wrote following a call with management. “The company is also getting feedback that there may be more read-through from what happens in the EU in terms of value-based pricing than one might anticipate.”
Analysts also eagerly await an update from Bluebird at next month’s European Hematology Association Congress in Amsterdam, where the company plans to present updated results up to 54 months from the long-term follow-up of the completed phase I/II Northstar study of Zynteglo in TDT. The company also expects to present data from the ongoing phase III Northstar-2 study in those who do not have a B0/B0 genotype and from the ongoing phase III Northstar-3 study in those with the B0/B0 genotype or an IVS-I-110 mutation.
Taking ‘a wait and see attitude’
In the U.S., where Bluebird expects to file for regulatory approval of betibeglogene darolentivec in beta-thal by year-end, the company’s foray into the indication also has produced significant interest among the patient and provider communities.
“I wouldn’t be surprised if a fair amount of patients have a wait and see attitude right now,” Neufeld said. “A commercial approval and then news of commercial success, if that occurs, is almost guaranteed to generate more interest.”
Other biopharmas have wasted little time following Bluebird’s lead. Twenty-eight assets, half of them in the clinic, are cited by Cortellis Competitive Intelligence, and differing mechanisms of action offer multiple treatment approaches in the space. (See chart, below.)
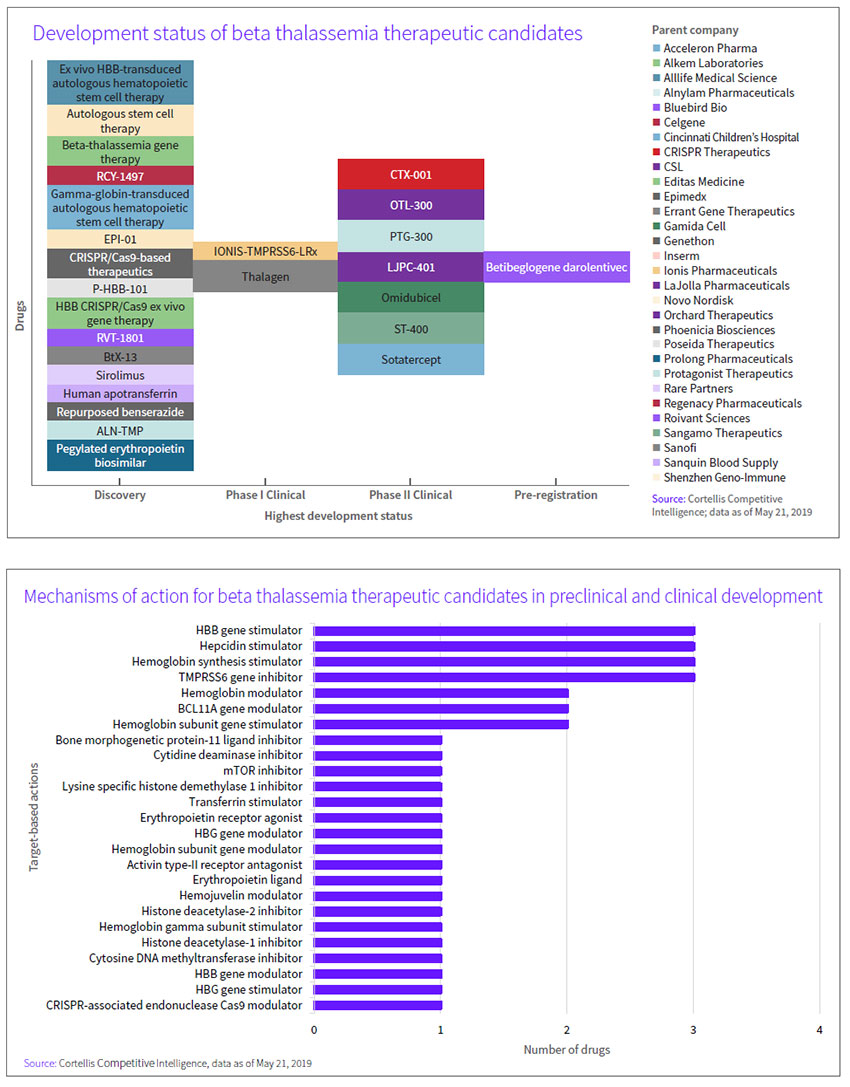
One alternative to adding a beta-globin transgene is to restart the endogenous gamma-globin gene that regulates the production of hemoglobin during fetal development, Neufeld pointed out. Several mechanisms of that kind, designed to knock down BCL11A, already are in the clinic. Among the options are ST-400, a BCL11A gene modulator from collaborators Bioverativ Inc., a unit of Sanofi SA, of Paris, and Sangamo Therapeutics Inc., of Richmond, Calif. The autologous stem cell therapy uses a patient’s CD34-positive stem cells, modified ex vivo using zinc finger nuclease transcription factors, to disrupt the BCL11A erythroid specific enhancer and promote the production of fetal hemoglobin. Last month, initial data from the first patient in phase I/II THALES study showed neutrophil and platelet recovery, within two and four weeks of infusion, respectively, indicating successfully reconstituted hematopoiesis following conditioning. Small insertions or deletions generated at the targeted DNA sequence were detected in circulating white blood cells, suggesting successful editing of the BCL11A gene and disruption of the BCL11A erythroid specific enhancer.
A competing effort is underway by partners Crispr Therapeutics AG, of Zug, Switzerland, and Vertex Pharmaceuticals Inc., of Boston. Subsequent to their potential $2.6 billion-plus genetic disease deal in 2015, last year the companies moved CTX-001 into separate phase I/II studies in TDT and SCD. The therapy consists of a single dose of autologous hematopoietic stem cells, genetically engineered ex vivo using CRISPR/Cas9, to disrupt the BCL11A erythroid specific enhancer and promote production of fetal hemoglobin. (See BioWorld Today, Oct. 27, 2015.)
Orchard Therapeutics Ltd., of London, accelerated its gene therapy prospects last year by taking the approved therapy Strimvelis and the rest of Glaxosmithkline plc’s rare disease gene therapy portfolio, including OTL-300, an ex vivo gene therapy targeting beta-thal that consists of autologous CD34+ hematopoietic stem/progenitor cells genetically engineered using a lentiviral vector, GLOBE, to express human beta-globin. The phase I/II proof-of-concept TIGET-BTHAL program has fully enrolled 10 participants, with data expected this year. Orchard raised $113.5 million in 2017 in advance of the gene therapy acquisition and another $150 million afterward to fund development of its enlarged portfolio. (See BioWorld, Dec. 20, 2017, April 13, 2018, and Aug. 14, 2018.)
Aruvant Sciences Holdings Ltd. also has a stake in the game. The company was launched last year by Roivant Sciences Ltd., in partnership with Cincinnati Children’s Hospital Medical Center, to advance RVT-1801, a lentivirus-based gene therapy that delivers the gamma-globin gene encoding fetal hemoglobin, though efforts for now are focused more on the treatment of SCD.
Ionis Pharmaceuticals Inc., of Carlsbad, Calif., completed a phase I trial and plans to open a phase II proof-of-concept study of IONIS-TMPRSS6-LRx, a TMPRSS6 expression-targeting ligand-conjugated antisense oligonucleotide, in beta-thal intermedia. Phase I data, presented in December at the ASH annual meeting in San Diego, showed mean transferrin saturation, reticulocyte hemoglobin and iron were reduced from baseline to day 141, while plasma hepcidin levels increased over the same period, with no serious adverse events (AEs) or treatment-emergent AEs.
Acceleron Pharma Inc., of Cambridge, Mass., and partner Celgene Corp., of Summit, N.J., already proved the mettle of red blood cell maturation agent luspatercept to treat beta-thal-related anemia in the phase III BELIEVE trial and, in April, filed a BLA with the FDA for the treatment of myelodysplastic syndromes and beta-thal-associated anemia requiring red blood cell transfusions. The companies also eyed an earlier-stage asset, sotatercept, a fusion protein containing a soluble form of the activin IIa receptor that inhibits signaling of the receptor and an Fc fragment of IgG1 to promote red blood cell production, for beta-thal, but current sotatercept development efforts appear aimed more at treatment of pulmonary arterial hypertension.
Protagonist Therapeutics Inc. originally considered out-licensing PTG-300, a disulfide-rich peptide mimetic of hepcidin developed using its Vectrix technology, to treat chronic anemia and iron overload in individuals with low hepcidin levels, including those with beta-thal. But in January, the Newark, Calif.-based company opened a phase II trial in those with anemia associated with non-transfusion-dependent or TDT. Data are expected by year-end.
‘The optimum strategy isn’t known’
Academic researchers around the globe are working on additional gene editing and gene therapy strategies in beta-thal, but whether a first approval to treat the underlying disease will generate more dollars for research remains to be seen.
“At present, all of the research efforts pique my interest,” Neufeld acknowledged. “They all have something to offer. The optimum strategy isn’t known. We’re just starting to see efforts to use CRISPR to aim for the erythroid specific enhancer of BCL11A or to aim for the BCL11A binding site – fetal globin promoters – but there’s not yet a clear path to success using CRISPR in human cells. The lentiviral approaches, the retroviral approaches to drive BCL11A knockdown and the genetic editing efforts all are appealing in their own way. It’s a very exciting time. Personally, I wouldn’t bet on one approach over the others.”
But even with the first of these new mechanisms on the cusp of commercial use, concerns about cost persist. In January, during the 37th Annual J.P. Morgan Healthcare Conference in San Francisco, Bluebird CEO Nick Leschly made the case for capped, at-risk, term-limited, installment-plan payments for potentially curative gene therapy and other one-time treatments. Patients and payers would pay only when a treatment works, with Bluebird at risk for up to 80% of the price, he said at the time. Payments would be spread over a period of up to five years, with no cost for the therapy beyond that payment period. Bluebird also committed to no price increases for future products beyond the consumer price index.
The company pegged the value of its lentiglobin-based product in the range of $3 million to $4 million. Cost offsets returned to the system would lower that “intrinsic” value to $2.1 million, which Leschly said “is not the price but does provide an upper bound” of the company’s decision-making on pricing the launch in Europe. The actual amount will be disclosed following formal approval, according to Bluebird, and be consistent across EU countries. (See BioWorld, Jan. 10, 2019.)
Although the proposal curried favor with a number of stakeholders, such as biopharma CEOs, payer representatives, key opinion leaders, patient advocacy organizations and government officials around the world, many still worry whether treatment will be affordable to those who need it most.
To mitigate anemia, Individuals with TDT typically require at least monthly blood transfusions. They also need chronic iron chelation therapy to prevent iron overload. Transfusion-chelation treatment for TDT in the U.S. and developed countries “is no bargain” for health care systems, at a cost of approximately $100,000 per year, or more than $1 million over a decade of treatment, Neufeld acknowledged. An alternative approach that’s curative would offset that cost.
Still, “I’m quite concerned about pricing for fancy new therapies,” he admitted. “Nowadays, with drug pricing in the U.S. and the developed world, people tend to want to charge many years’ worth of conventional therapy up front for a successful new treatment. I’m somewhat a critic of that approach.”
Even if there aren’t enough individuals with severe TDT in the U.S. to break the bank at a charge of $2 million up front for gene therapy, the cost on a per-patient basis “is pretty extreme,” Neufeld said. “We need to look at payment models that enable drug companies to succeed without charging so much.”
The stakes are even higher, he said, in countries such as Thailand, where tens of thousands of people have TDT and a one-time gene therapy priced 10 to 20 times more than the cost of annual treatments is not sustainable.
“I understand that companies have a commercial interest, so it’s a challenge,” Neufeld said. “But aiming for very high prices because a company can do that doesn’t mean it’s a good idea.
“Whether patients who are poor can get these treatments at cost is a question that interests me,” he added. “In drug development, we don’t often know the cost of goods. But in the commercial gene therapy world, the cost of goods is terribly important because it determines how many people you can potentially cure for a given amount of money. That’s not a consideration people often ponder.”
